



分享:Al摻雜Mg/Mg2Sn合金界面的第一性原理計算

王福容1, 張永梅1, 柏國寧2, 郭慶偉2, 趙宇宏 ,2,3
,2,3
1
2
3
為研究Mg-Sn合金中Al摻雜Mg基體與Mg2Sn相不同取向以及Al元素在界面處的分布位置,基于密度泛函理論計算了Al元素摻雜Mg/Mg2Sn不同指數面的界面黏附功、界面能以及界面摻雜能來尋找較穩定的摻雜位置。采用態密度和晶體軌道重疊布居分析了Al元素摻雜對Mg(0001)/Mg2Sn(022)界面電子特性的影響。結果表明,界面處添加Al元素后只有部分摻雜位置有益于加強Mg/Mg2Sn界面的穩定性。添加Al元素后,Mg(0001)/Mg2Sn(001)界面處Sn端黏附功均高于Mg端,而Mg(0001)/Mg2Sn(111)界面正好相反。Al摻雜后的Mg(0001)/Mg2Sn(022)界面能降低了0.07 eV/nm。添加Al元素后,Mg(0001)/Mg2Sn(022)界面位置Ⅳ比較容易摻雜,該位置處的電子結構分析表明摻雜Al元素后Al的s軌道和Sn的p軌道存在明顯交互作用,在界面處Al—Sn鍵占主導地位。
關鍵詞:
鎂合金因其質量輕、資源豐富以及比強度/比剛度高而廣泛應用于航空航天、汽車等領域[1~3]。Mg-Sn合金成本低,具有反螢石結構的Mg2Sn沉淀相的沉淀硬化效應顯著[4~6],也可以改善合金抗蠕變性能[7,8]。但Mg2Sn相會快速長大并粗化,從而惡化Mg-Sn合金的力學性能。
通過微合金化可以減少Mg2Sn的析出從而提高合金的性能,這得益于其固溶強化效應[9,10]。Sasaki等[11]研究發現,添加Zn元素可以改變Mg2Sn和Mg基體之間的界面能,有利于細化Mg2Sn沉淀物。Mendis等[12]將Mg2Sn作為模型,分別添加Na和In + Li,發現添加Na使得硬化增量比添加In + Li增加了近一倍。Pan等[13]研究證實添加Ca可以在Mg-Sn合金中形成熱穩定相CaMgSn,之后添加Y和Zr可以細化CaMgSn相,從而提高合金強度和延展性。田樹科和郭學鋒[14]發現在Mg-Sn合金中分別添加Al和Zn元素時,都可以提高合金的固溶強化效果,Al的固溶強化效果高于Zn。Kim和Park[15]發現Al元素的加入可以改善Mg-Sn合金的力學性能和硬度。Luo等[16]證實了Al的添加可以顯著改變析出相Mg2Sn的分布和形貌,并對Mg2Sn起到細化作用,改善Mg-Sn合金的力學性能。徐孝新[17]發現在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基體與Mg2Sn界面處,降低Mg2Sn的界面能,減小形核功,使形核過程更易于進行。然而實驗上難以從微觀角度分析界面結合機制,需要結合第一性原理[18~20]、分子動力學等方法進行分析。目前第一性原理在研究合金化元素改善界面性質方面取得了很多成果[21~24]。王小宏等[25]基于第一性原理研究了Al、Zn占位對Mg/Li相界斷裂強度的影響,表明Al、Zn元素的添加提高了體系穩定性。Liu等[26]預測了Cr、Os、Ir元素摻雜對NiCo/Cu界面的結合強度和力學性能的影響,并通過原子位置、鍵長和電子性質解釋了加強機理。Zhang等[27]通過第一性原理發現了Sc在S1位點摻雜AlCu/Al界面會降低其界面能,并且增強其黏附功,特別是被間隙Cu原子占據的S1位點與Sc的摻雜具有非常好的結合強度。在Mg-Sn合金中,粗化的Mg2Sn相會導致該合金的時效硬化效果降低,而Al元素可以顯著提高Mg-Sn合金的時效硬化效應。在實驗中觀測到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基體與Mg2Sn相的界面,起到細化作用。第一性原理可以定性給出界面處的電子結構以及合金元素對界面穩定性[28,29]的影響,但對于Al摻雜Mg/Mg2Sn界面性質的研究很少。
本工作對Mg/Mg2Sn界面進行討論,從原子尺度研究了在不同界面上的不同位置摻雜Al元素對Mg/Mg2Sn界面性能的影響,并從電子層次解釋了Al摻雜前后Mg(0001)/Mg2Sn(022)界面處原子軌道變化,分析了Al合金化提高Mg-Sn合金性能的影響機理。
1 模型和方法
1.1 模型構建
Mg/Mg2Sn界面結構基于hcp結構的Mg基體[30]以及fcc結構的Mg2Sn析出相[31]建立。對于Mg基體結構,晶格常數a = b = 0.320 nm,c = 0.513 nm;α = β = 90°,γ = 120° (α、β和γ分別為晶格常數a和b、a和c、b和c 3個軸之間的夾角)。Mg2Sn析出相為fcc結構,a = b = c = 0.681 nm,α = β = γ = 90°。有實驗觀察到Mg2Sn與Mg基體之間的取向關系為
表1 Mg/Mg2Sn界面間距以及界面能和晶格錯配度
Table 1
| Interface structure | Interface spacing / nm | Interface energy / (eV·nm-1) | Mismatch degree (δ) / % |
|---|---|---|---|
| Mg(0001)/Mg2Sn(001)-Mg | 0.350 | 18.14 | 6.34 |
| Mg(0001)/Mg2Sn(001)-Sn | 0.350 | 31.52 | 6.34 |
| Mg(0001)/Mg2Sn(022) | 0.250 | 14.58 | 3.05 |
| Mg(0001)/Mg2Sn(111)-Mg | 0.190 | 17.56 | 2.56 |
| Mg(0001)/Mg2Sn(111)-Sn | 0.190 | 27.29 | 2.56 |
比較3個不同取向界面能可知,Mg(0001)/Mg2Sn(022)界面能最小,界面較穩定。為了研究Al摻雜位置對界面穩定性的影響,對于不同取向界面分別在Mg(0001)表面以及Mg2Sn表面找尋了不同的Al摻雜位置,如圖1中藍色虛線球所示。圖1a和b分別為Mg(0001)/Mg2Sn(001)界面的Mg端界面和Sn端界面;圖1c為Mg(0001)/Mg2Sn(022)界面;圖1d和e分別為Mg(0001)/Mg2Sn(111)界面的Mg端和Sn端。其中紫色球代表Sn原子,橘色球代表Mg原子,藍色虛線球代表摻雜的Al原子,綠色虛線框代表Mg/Mg2Sn界面,羅馬數字代表界面處Al元素的摻雜位置。
圖1

圖1 Al元素摻雜不同界面取向的不同位置的Mg/Mg2Sn界面模型示意圖
Fig.1 Schematics of Mg/Mg2Sn interface model with different interface orientations doped with Al element (Roman numerals represent the different doping positions of Al at the interface) (a, b) Mg terminal (a) and Sn terminal (b) models of Mg(0001)/Mg2Sn(001) interface, respectively (c) Mg(0001)/Mg2Sn(022) interface model (d, e) Mg terminal (d) and Sn terminal (e) models of Mg(0001)/Mg2Sn(111) interface, respectively
1.2 計算方法
本工作所有計算均使用基于密度泛函理論的VASP[32]軟件包進行。選用Perdew-Burke-Ernzerhof (PBE)的廣義梯度近似(generalized-gradient-approximation,GGA)為電子交換關聯泛函。經過收斂性測試,平面波截斷能為400 eV。Monkhorst-Pack方案的k點最終設定為9 × 9 × 4和5 × 5 × 5用于Mg和Mg2Sn進行優化計算,4 × 4 × 1、5 × 3 × 1和3 × 2 × 1分別用于Mg(0001)/Mg2Sn(001)、Mg(0001)/Mg2Sn(022)和Mg(0001)/Mg2Sn(111)界面的計算。采用4 × 5 × 1、4 × 4 × 1、4 × 3 × 1和3 × 2 × 1分別對4層Mg2Sn(001)、4層Mg(0001)、4層Mg2Sn(022)以及4層Mg2Sn(111)表面進行計算。收斂參數設置:電子弛豫的收斂標準為10-5 eV,并且對于界面計算,結構優化收斂標準為結構內任何原子的作用力均小于0.1 eV/nm。考慮到表面原子間的相互作用,沿Z軸方向設置了長度為1 nm的真空層,使得2個相鄰界面之間的相互作用可以忽略。
2 計算結果與討論
2.1 界面黏附功
黏附功(Wad)反映了界面結構與2個表面結構之間的能量差[32]。Wad可以反映界面結構的力學穩定性以及抵抗裂紋擴展的能力。穩定的界面結構對應于高的Wad。它被定義為單位面積上將界面分離成2個自由表面所需的能量[33]:
式中,
圖2
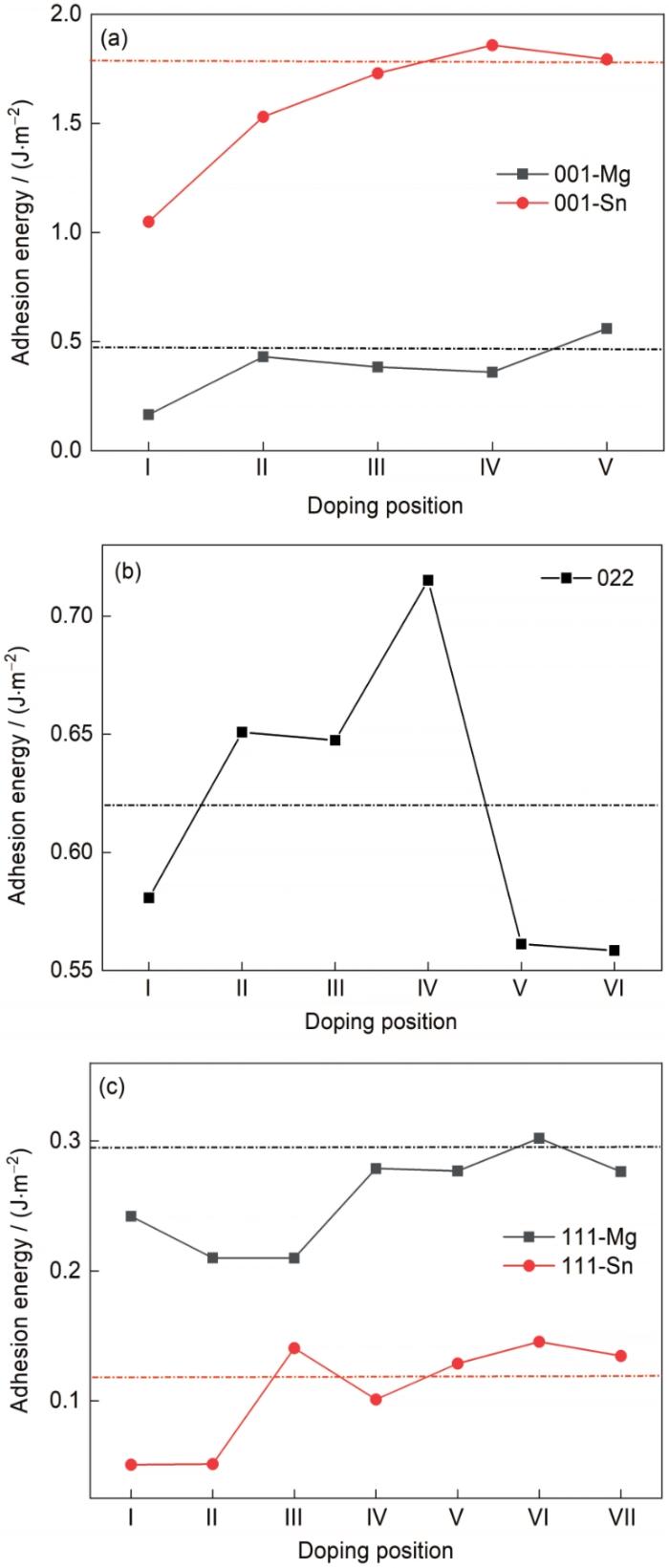
圖2 Mg/Mg2Sn界面不同位置摻雜Al元素的黏附功
(a) Mg(0001)/Mg2Sn(001) interface
(b) Mg(0001)/Mg2Sn(022) interface
(c) Mg(0001)/Mg2Sn(111) interface
Fig.2 Adhesion energies of Mg/Mg2Sn interface with doping Al element at different positions (Dotted lines represent the interface adhesion energies without Al element)
2.2 界面摻雜能
界面摻雜能(Ef)可以反映摻雜界面形成的難易程度,其值越小越容易摻雜[34]:
式中,
表2 Al摻雜Mg(0001)/Mg2Sn(022)界面的界面間距、界面能和摻雜能
Table 2
| Position | Interface spacing / nm | Interface energy / (J·m-2) | Interface doping energy / (J·m-2) |
|---|---|---|---|
| Ⅰ | 25.84 | 0.418 | -5.031 |
| Ⅱ | 23.64 | 0.143 | -5.070 |
| Ⅲ | 23.20 | 0.140 | -5.076 |
| Ⅳ | 22.83 | 0.091 | -5.079 |
| Ⅴ | 23.22 | 0.149 | -5.075 |
| Ⅵ | 23.27 | 0.152 | -5.075 |
2.3 界面能
總的界面形成能包括界面能和彈性應變能。界面能是指Mg/Mg2Sn構建的界面體系分成獨立晶體Mg和Mg2Sn所需的能量,而彈性應變能是指Mg基體和Mg的析出相Mg2Sn之間所需的應變能。考慮到界面周圍最近原子層的影響,對Mg(0001)/Mg2Sn(022)界面進行擴胞,構建了原子個數分別為56、112和168的3個晶胞,計算總的界面形成能[35]:
式中,ΔGf為界面每個原子的界面形成能,γ為單位面積的界面能,ΔGcs為平均每個原子的彈性應變能,
圖3
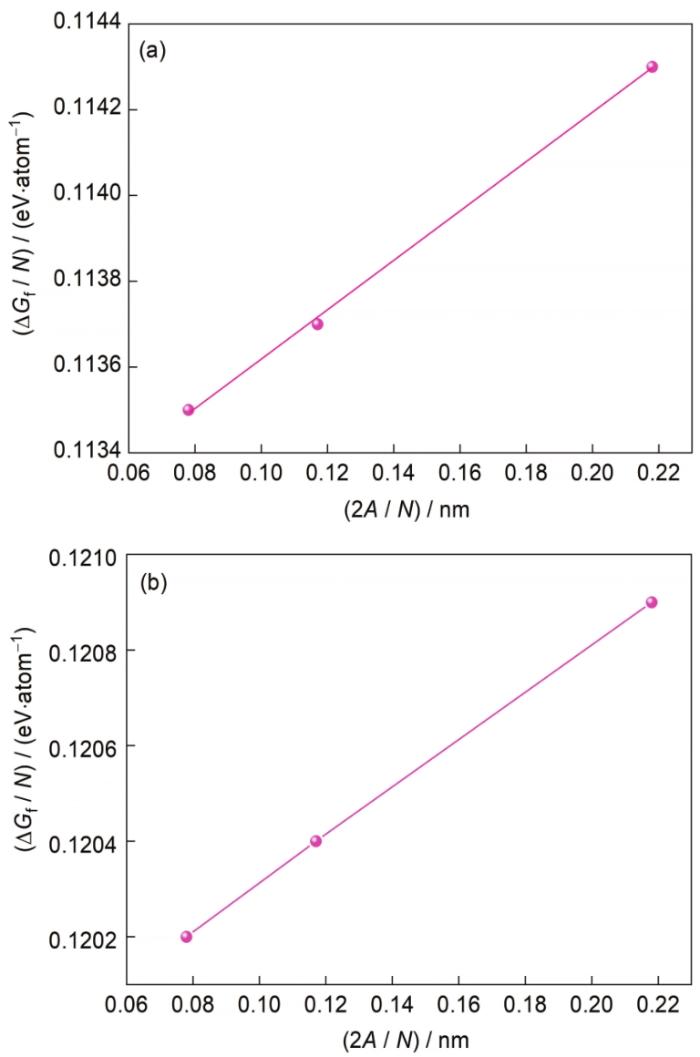
圖3 Mg(0001)/Mg2Sn(022)界面位置Ⅳ摻雜Al元素前后的界面能
Fig.3 Interface energies of Mg(0001)/Mg2Sn(022) before (a) and after (b) doping Al element at position IV in Fig.1c (The intercept of the line in the figure represents the elastic strain energy, and the slope represents the interface energy. A—interface area, N—number of atoms in the interface, ΔGf—energy contained in a single atom in the interface)
2.4 界面電子特性
為了更深入地了解Mg(0001)/Mg2Sn(022)界面處摻雜Al原子的成鍵特性,繪制了摻雜Al元素前后Mg(0001)/Mg2Sn(022)界面的分波態密度(partial density of states,PDOS)曲線,如圖4所示,圖中黑色虛線代表Fermi能級(EF)。分波態密度可以反映出界面處摻雜元素前后界面結構穩定性差異的物理本質[36]。從圖4a可以看出,第1~3層Mg和Sn在-4~0 eV處存在明顯的重疊波峰,2者間存在較強的雜化共軛作用,界面處主要是Mg的s、p軌道與Sn的p軌道之間的相互作用,導致Mg、Sn原子之間形成較強的共價鍵。將圖4a與b對比后發現,在界面第1層處添加Al元素后Mg、Sn、Al不僅在-4~0 eV處存在明顯的重疊波峰,在-6.4~-4 eV處也出現了重疊波峰,并且Al的s軌道和Sn的p軌道存在明顯的軌道雜化作用,使得Al摻雜界面的結合作用更強,結構更穩定。
圖4
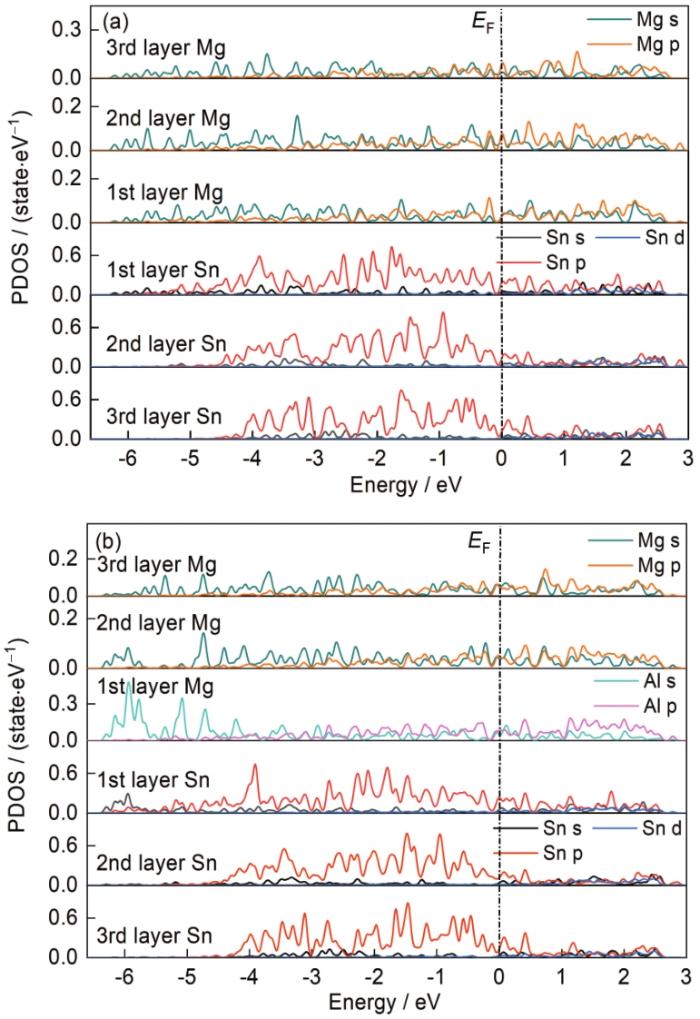
圖4 Mg(0001)/Mg2Sn(022)界面位置IV摻雜Al元素前后的分波態密度(PDOS)曲線
Fig.4 Partial density of states (PDOS) curves of Mg(0001)/Mg2Sn(022) interface before (a) and after (b) doping Al element at position IV in Fig.1c (EF—Fermi level)
圖5為摻雜Al前后Mg(0001)/Mg2Sn(022)界面處差分電荷密度差異。可以發現,Mg(0001)/Mg2Sn(022)界面上存在明顯的電荷轉移,在界面的形成過程中電子結構重新排列,界面原子周圍出現電荷的聚集和損耗,紅色區域代表電荷的聚集,藍色電荷代表電荷的損耗,說明形成了化學鍵。從圖5a1和a2可以發現Mg(0001)/Mg2Sn(022)界面處Mg原子周圍是藍色區域,說明Mg原子失去電子,而電子均流向電負性比較強的Sn原子處,Sn原子周圍是紅色區域,實現了電荷的大量聚集,故在界面處形成Mg—Sn共價鍵。從圖5b1和b2可以發現,摻雜Al之后,界面處Al原子周圍藍色區域更大,失電子更多,這表明界面摻雜Al原子之后界面結合強度增強。
圖5

圖5 Mg(0001)/Mg2Sn(022)界面位置IV摻雜Al元素前后的差分電荷密度圖的側視圖和俯視圖
Fig.5 Side views (a1, b1) and top views (a2, b2) of differential charge density diagrams of the Mg(0001)/Mg2Sn(022) interface before (a1, a2) and after (b1, b2) doping Al element at position IV in Fig.1c (The red areas represent the accumulation of electric charge, and the blue areas represent the loss of electric charge)
基于對圖5分析可以發現結構中存在廣泛的共價鍵,但缺乏對這些共價鍵準確定量測量以及鍵強度和貢獻的分析。晶體軌道Hamilton布居(crystal orbital Hamilton populations,COHP)可以定量分析晶體結構當中2個原子的結合強弱,而晶體軌道重疊布居積分值(integrated crystal orbital Hamilton populations,ICOHP)對應于Fermi能級積分能量值[37]。圖6a和b分別為hcp結構Mg和fcc結構Mg2Sn的投影晶體軌道Hamilton布居(projected crystal orbital Hamilton populations,pCOHP)[38]曲線,圖6c~f為Mg(0001)/Mg2Sn(022)構建的界面以及在界面處摻雜Al的2種界面在界面處的Mg—Mg、Mg—Sn原子的pCOHP曲線。圖中右側的峰值(-pCOHP > 0)代表的是各個軌道成鍵貢獻,左側的峰值(-pCOHP < 0)代表的是各個軌道反鍵態的貢獻。在這些原子相互作用中,比較圖6c、d以及圖6e、f,不難發現在界面處摻雜Al原子后,界面處Mg—Al之間以及Al—Sn之間都存在相互作用,主要是Mg3p、Al3p以及Sn3p、Al3p提供成鍵軌道。對于ICOHP的值排列如下:Mg3s—Sn5p > Mg3p—Al3p > Mg3s—Mg3p > Mg3p—Mg3p > Mg3p—Sn5s > Al3p—Sn5p,在界面處Al—Sn鍵強度占主導地位,Mg—Al鍵次之。通過將界面處原子之間相互作用分解為對不同軌道的貢獻,對于不摻雜Al的界面,界面處Mg—Mg之間主要是3p軌道提供成鍵,Mg—Sn之間主要是Mg3p、Sn5s、Sn5p提供成鍵。
圖6

圖6 hcp結構Mg和fcc結構Mg2Sn,Mg(0001)/Mg2Sn(022)界面處的Mg—Mg和Mg—Sn原子,以及摻雜Al后的Mg(0001)/Mg2Sn(022)界面處的Mg—Al和Al—Sn原子的投影晶體軌道Hamilton布居(pCOHP)曲線
Fig.6 Projected crystal orbital Hamilton population (pCOHP) curves of hcp Mg (a) and fcc Mg2Sn (b); Mg—Mg (c) and Mg—Sn (d) atoms at the Mg(0001)/Mg2Sn(022) interface; and Mg—Al (e) and Al—Sn (f) atoms at the Mg(0001)/Mg2Sn(022) interface after doping Al (ICOHP—integrated crystal orbital Hamilton population)
3 討論
在實驗研究中,Al元素是改善鎂合金力學性能的主要元素之一。有不少研究證實了隨著Mg-Sn合金中Mg2Sn的不斷析出,Mg2Sn相會粗化,導致合金力學性能變差。添加Al元素可以抑制合金內Mg2Sn析出相的粗化,對Mg2Sn起到細化作用,提高合金的力學性能[14~16]。然而實驗上只是在宏觀上分析了Al的添加改善了Mg-Sn界面的力學性能,相比于實驗上只是觀測到Al摻雜有益于提高Mg/Mg2Sn界面的穩定性,本工作從原子尺度分析解釋了摻雜Al元素對Mg/Mg2Sn 3種不同取向界面穩定性的影響,以及界面處不同摻雜位置對界面性質的影響,發現摻雜位置會影響Mg/Mg2Sn界面的穩定性,有的摻雜位置對提高界面的穩定性并不是有益的。Al摻雜后的Mg(0001)/Mg2Sn(022)界面能降低了0.07 eV/nm,這與徐孝新[17]在實驗上觀察到在Mg-Sn合金中加入Al元素后Al偏聚在Mg基體與Mg2Sn界面,導致界面能降低的結果一致。并且通過對Al摻雜Mg(0001)/Mg2Sn(022)界面電子特性的分析,界面處Al—Sn鍵強度占主導地位,解釋了實驗上觀測到的Al元素的加入增強了Mg-Sn界面的結合強度。
4 結論
(1) 在Mg(0001)面和Mg2Sn 3個低指數面的不同位置添加Al元素后,尋找到較穩定的Al元素摻雜位置。對于Mg(0001)/Mg2Sn(001)界面,Mg端界面所選5個位置在位置Ⅴ添加Al元素能加強界面的結合強度;對于Sn端,Sn端的界面黏附功遠遠高于Mg端,在位置Ⅳ和位置Ⅴ添加Al元素都可以加強界面的穩定性。對于Mg(0001)/Mg2Sn(022)界面,在位置Ⅱ、III、Ⅳ添加Al元素都可以加強界面的結合強度,其中位置Ⅳ黏附功比其他位置高。對于Mg(0001)/Mg2Sn(111)界面,界面處Mg端所選Al元素摻雜位置的黏附功都高于Sn端,說明Mg終端更利于界面的穩定性。
(2) 在Mg(0001)/Mg2Sn(022)界面6個位置摻雜Al,其界面摻雜能都為負值,位置IV最容易摻雜Al元素,位置I最不易摻雜。Mg(0001)/Mg2Sn(022)界面能為0.37 eV/nm (0.593 J/m2),摻雜Al后的界面能為0.30 eV/nm (0.481 J/m2),添加Al元素后界面能變小,界面變穩定。
(3) 分波態密度以及差分電荷密度分析結果表明,界面處加入Al元素后,Al的s軌道和Sn的p軌道存在明顯的相互作用。界面處Al原子周圍藍色區域更大,失電子更多,在界面處Al—Sn鍵強度占主導地位。
來源--金屬學報 滬公網安備31011202020290號
滬公網安備31011202020290號
