



分享:水分子在Au和Cu及其合金表面的吸附與分解DFT計算研究

蔣宗佑
摘要
采用密度泛函方法研究了水分子在Au, Cu和AuCu二元金屬合金表面的不同吸附狀態和裂解反應路徑, 并且比較不同表面的催化性能. 結果表明, 研究中所考慮4種模型的反應活性順序如下(以反應活化能為比較標準): Au(111) < AuCu(111)-Cu < AuCu(111)-Au < Cu(111). 相對于AuCu(111)-Cu表面和Au(111)表面, 這與水分子的分子吸附狀態在AuCu(111)-Au表面和Cu(111)表面的吸附能相對較小, 而解離吸附狀態時的吸附能相對較大有關. 根據金屬催化劑表面與吸附物的電子轉移和成鍵電子結構, 認為電子轉移越多, 與表面的相互作用越強; 而金屬原子的d電子態與水分子1b1電子態或者裂解產物的類1b1電子態之間的重疊、雜化程度決定了兩者之間相互作用的強弱. 在實際反應體系中, 用AuCu二元金屬合金替代貴金屬Au催化劑不僅可以降低材料成本, 還可以提高反應活性.
關鍵詞:
Au作為一種具有化學惰性的貴金屬, 廣泛應用于珠寶首飾及微電子集成電路等領域. 作為一種催化劑, 對Au的研究最早可追溯到1926年Au催化H2+O2的反應, 但是Au的催化反應速率比Ag的要低得多[1]. 1985年, Hutchings[2]在研究乙烯氫氯化反應機理時, 曾預測負載型Au催化劑是該反應中催化活性最高的催化劑, 但是這一結論直到1988年才被實驗證實. 1987年, Haruta等[3,4]報道超細Au顆粒顯示出極高的CO低溫氧化活性. 雖然Au的催化活性較低, 但是其本身具有很高的催化選擇性[5], 因此Au催化劑大多數情況下應用于選擇性氧化反應和有機反應.
Au在催化領域中最主要的3種應用形態是: 負載型Au顆粒、Au薄膜和Au納米顆粒. 2015年, Wu等[6]發現高價氧化態的Au3+有極強的親氧能力, 可取代強氧化劑, 并且其前驅體很容易制備且易于保存, 這是催化領域中的一個重大發現. 喬波濤等[7]通過簡單的沉淀吸附法制備Co3O4負載Au單原子催化劑(Au1/Co3O4), 發現Au負載量僅為0.05% (質量分數)即可在室溫條件下實現CO的完全轉化, 并提出了“單原子催化”的概念, 表明負載型Au單原子在CO氧化方面具有獨特的催化作用. 除了自身具有的催化活性之外, 納米Au粒子還可以作為助催化劑負載在光催化材料表面, 可增加光生電子-空穴對的分離效率, 從而提高光催化材料的活性. 例如, 代衛炯等[8]用光沉積法在TiO2載體上負載Au, 發現在Au和TiO2界面處形成的Schottky勢壘, 有效阻止了光生電子與空穴的復合. 相比于沒有Au助催化劑存在的情況, 前者的光催化活性得到了大幅提高. 由于負載型Au催化劑的獨特性能, 負載型Au催化劑催化CO低溫氧化的反應仍然是目前的研究重點. 但是, Au本身價格昂貴, 資源有限, 且其催化活性也有待提高, 因此需要引入新的思路, 在保持其催化活性的前提下減少Au的用量.
為了降低催化劑成本并盡可能提高催化活性, 在實際應用中通常考慮以非貴金屬替代部分貴金屬. 相關研究[9~12]表明, 過渡金屬Cu是最有希望的非貴金屬催化劑之一. 在催化領域中, Cu主要應用在有機反應中的催化方面, 包括催化偶聯反應、催化多組分反應和Cu金屬配合物等應用. 1901年, Ullmann和Bielecki[13]首次報道了Cu催化的交叉偶聯反應, 即2個芳基直化物之間形成C—C鍵的偶聯反應, 開創了Cu在催化偶聯反應方面的先河. Nadler和Sanz[14]采用第一性原理分子動力學模擬H2O在Cu(111)表面的作用, 發現Cu表面的存在將改變H鍵網絡, 并使H—O鍵加強, 這種影響類似于將純H2O加熱所帶來的結果. 2016年, Shao等[15]報道了在Cu催化不對稱炔丙基轉化研究中, 通過運用一種脫硅活化的新方法, 成功實現了Cu催化的炔丙醇酯與β-萘酚及富電子苯酚間的不對稱[3+2]環加成反應. 2009年, 厲嘉云等[16]報道了N-雜環卡賓Cu配合物的應用前景及其催化機理. 由于納米技術的迅猛發展, 研究人員發現包括Cu及其化合物在內的納米材料具有很多新的特點, 比如催化活性高、應用范圍廣、環境友好、價格低廉、能多次循環利用等. 2014年, Manthiram等[17]發現將Cu納米粒子負載于玻璃碳上面(n-Cu/C)并用于CO2的甲烷化反應, 其效率比單獨使用高純度Cu箔電極高了4倍. Kundu和Pradhan[18]發現納米結構的CuS對亞甲基藍降解有著較高的效率. 此外, 工業上的骨架Cu催化劑主要作為丙烯腈水合制丙烯酸胺反應的催化劑, Cu系催化劑在CO轉化及某些典型的加氫還原反應中具有較高的活性. 鑒于Cu在催化領域的諸多優點, 以及其成本的低廉與儲量的豐富, 研究者希望找到一種將Au的特點與Cu的優點相結合的有效途徑.
研究[19]發現, 金屬與金屬之間形成的雙/多金屬結構, 其物理化學性能往往優于單金屬, 這種現象被稱之為金屬之間的協同效應. 由于催化劑的表面結構是決定其催化性能的關鍵因素, 而引入第二種金屬又可以改變單金屬作為催化劑時的表面結構, 因此可以將2種或多種金屬通過不同比例、不同結構與不同組合的方法制備成具有較好催化性能的雙/多金屬催化劑. 而Au-Cu作為貴金屬體系中重要的基礎合金體系, 在催化領域中, 不僅降低了Au單獨作為催化劑時的成本, 也提高了Cu或Au單獨存在時的催化性能. 2015年, Fiorenza等[20]在揮發性有機化合物的氧化和CO選擇氧化實驗中發現, 在低溫下, Au-Cu/CeO2比Au/CeO2或Cu/CeO2擁有更高的CO2產量. 2012年, 李力成等[21]制備出負載型雙金屬催化劑Au-Cu/TiO2, 并用于CO催化氧化反應中. 發現Cu可以提高Au催化劑的催化性能, Au-Cu/TiO2催化劑比Au/TiO2和無孔TiO2負載的Au催化劑具有更好的CO催化氧化穩定性. 這可能與Au, Cu合金化和TiO2的介孔結構有關.
盡管關于Au-Cu合金催化劑的實驗報道較多, 但對其相關的理論研究還比較少, 迫切需要從理論上研究其催化反應的機理, 以解釋現有的實驗現象并指導后期的研究工作. 不論是均相催化、非均相催化或者其它類型的催化反應, 催化過程大都發生在催化材料的表面, 所以反應物分子的吸附分解是催化反應的必經步驟, 即催化反應的機理與反應分子在催化劑表面的吸附行為是不可分割的.
本工作以Au, Cu及其合金為研究對象, 通過理論模擬與計算, 揭示其催化的微觀機制. H2O是最常見的溶劑, 水分子本身就可以參與分解反應產生O2和H2, 且水分子結構簡單, 同時它可以通過自身的解離產物(如表面羥基、羥基自由基等是一些催化反應的活性物種)影響后續的催化反應, 因此將水分子作為研究催化機理的理想探針分子. 本工作從微觀角度, 通過理論計算, 模擬催化過程中H2O在Au, Cu和Au-Cu合金表面的吸附與分解, 研究其吸附狀態、成鍵機理和活性吸附位點等, 并據此分析Au, Cu和Au-Cu合金催化劑的反應機理.
本研究所有理論計算工作均由Materials Studio軟件中的CASTEP (Cambridge serial total energy package)模塊完成. 采用平面波超軟贗勢方法來描述電子-離子實間的相互作用, 以保證在一定計算精度的前提下節省計算時間和計算資源. 電子波函數通過平面波基組展開, 其中平面波截斷能設置為380 eV. 電子-電子間相互作用的交換關聯能由廣義梯度近似(generalized gradient approximation, GGA)中的PBEsol泛函進行描述, 這是目前對固體材料較為準確的理論計算方法. 幾何結構優化收斂標準設置如下: 2次相鄰離子步之間的總能量差小于1.0×10-6 eV/atom, 原子間相互作用力小于0.1 eV/nm, 原子所受應力小于0.02 GPa, 原子位移小于5.0×10-5 nm. 自洽場運算中電子步的能量收斂標準為5.0×10-7 eV/atom. 其它相關設置如下: K-points設置為2×2×1, 快速Fourier變換(FFT)的網絡設置為54×54×360. 為了得到更加準確的計算結果, 所有計算均采用偶極校正, 以消除表面不對稱所導致的影響.
Au和Cu的原始模型采用標準fcc結構(空間群為
對于水分子的分解過程, 本工作只分析對比裂解的初始階段, 即只有一個O—H斷裂的情形. 在對不同可能的分子吸附和解離吸附構型進行幾何結構優化、并得到最穩定的吸附構型之后, 以分子吸附作為反應物, 以解離吸附作為反應產物. 然后采用完全線性同步轉變/二次同步轉變(complete linear synchronous transit/quadratic synchronous transit, Complete LST/QST)方法搜索反應物和反應產物之間的過渡態, 其中: RMS收斂設置為0.1 eV/nm, 最大QST步數設置為5. 在得到初始過渡態之后再次進行過渡態確認, 其收斂標準設置為: 總能量差小于5.0×10-6 eV/atom, 原子間相互作用力小于0.2 eV/nm, 原子位移小于3×10-4 nm, 最大的鏡像數設置為10.
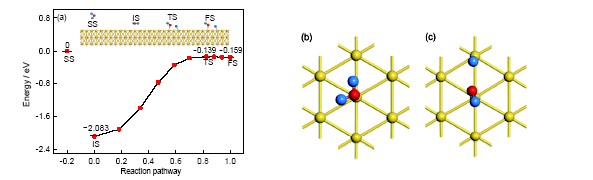
Au的密排面(111)晶面是其作為催化材料研究最多的對象[22]. 本計算中首先以水分子作為研究對象, 分析其在Au(111)晶面的吸附與分解行為, 計算結果如圖1a所示, 水分子在此表面上的分子吸附狀態和解離吸附狀態對應的吸附能分別為2.083和0.159 eV. 即從水分子到H+OH的分解過程是吸熱反應, 對應的反應熱為1.924 eV. 也就是說, 水分子在Au(111)晶面吸附時, 分子吸附狀態在能量上更占優勢, 在一般情況下水分子在此晶面上以分子吸附為主. 相關的結構參數列于表1. 結合表1和圖1可發現, 水分子在分子吸附狀態時, 水分子的分子平面與Au(111)晶面接近于平行(夾角約為4.375°), 2個O—H鍵的鍵長和鍵角與孤立水分子的鍵長和鍵角也基本相同, 只有稍許變化, 說明表面對水分子的微觀結構影響很小, 這可能與該表面上H2O的微觀結構比較穩定有關. 值得注意的是, 水分子在此狀態下, 2個H原子幾乎完全對稱地指向鄰近的2個Au原子, 這種相互作用使其保持了與孤立分子基本相同的結構. 解離吸附時H+OH基團的平面與Au(111)晶面的夾角是57.029°, 未裂解的O—H鍵長與孤立水分子中的O—H鍵長相差很小, 裂解之后的O—H鍵長為0.2237 nm. 裂解后產生的H原子與鄰近的Au原子成鍵, 兩者之間的鍵長為0.1619 nm (這一鍵長只相當于分子吸附時H原子與對應Au原子之間距離的一半). 在2個狀態之間反應路徑上的鞍點(即過渡態)對應的吸附能為0.139 eV, 在此狀態H+OH基團的平面與Au(111)晶面的夾角是74.141°. 在反應路徑上, 過渡態的結構是不穩定的, 它向前向后都很容易成為分子吸附或解離吸附狀態. 從圖1a中可以發現, 由于過渡態與終態(反應產物)存在一定的相似之處, 所以這一過渡態屬于“晚壘”. 從能量角度來看, 以分子吸附為初始態、解離吸附為終態的反應活化能為1.944 eV, 其逆反應的活化能為0.020 eV, 即在Au(111)晶面上水分子分解反應的逆反應更容易進行, 所以在此表面上水分子以分子吸附為主要的存在形態. 分子吸附與解離吸附的局部示意圖分別如圖1b和c所示. 可以看出, 2種吸附狀態都是頂位吸附, O原子與其所吸附的Au原子的距離分別為0.2571和0.2113 nm.
圖1 水分子在Au(111)表面上不同的吸附狀態與反應路徑, 及分子吸附和解離吸附的局部構型
Fig.1 Different adsorption states and reaction path of water on Au(111) surface (a), and the local configurations of molecular adsorption (b) and dissociation adsorption (c) (SS—separated state, IS—initial state, TS—transit state, FS—final state)
表1 本工作計算得到水分子在不同材料表面吸附與解離的結構參數
Table 1 Structural parameters of water adsorption and dissociation on different materials surfaces in this work
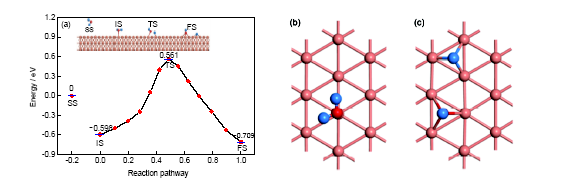
Cu(111)表面的催化性能也是受關注較多的課題之一[23,24]. 從表1及圖2可以看出, 水分子處于分子吸附狀態時, 情形與水分子在Au(111)表面的分子吸附狀態類似: 水分子的鍵長、鍵角與孤立分子基本一致, 對稱指向鄰近的2個Cu原子, 分子平面幾乎平行于表面. 但是在數值上有細微的差別, 如鍵長、鍵角稍大, 與表面的夾角相對較大(約為15.190°). 在裂解過程中, 水分子的分子平面逐漸豎立在表面上, 其中1個H原子被活化而與O原子分離, 吸附到鄰近的空位上, 而裂解形成的OH基團則以O原子端朝下吸附最近鄰的空位上, 成為表面吸附羥基, 形成最終的解離吸附狀態. 在解離吸附狀態下, H+OH基團的平面與表面幾乎垂直(夾角約為88.792°), OH基團的氧原子和裂解的H原子與周圍3個Cu原子分別形成等邊四面體結構, O—Cu鍵長為0.1977 nm, H—Cu鍵長為0.1735 nm. 從反應路徑上來看, 水分子在此表面上的解離吸附比分子吸附更占優勢, 對應吸附能分別為0.709和0.598 eV. 也就是說, 水分子在Cu(111)表面上為分子吸附狀態, 反應物的反應熱為0.111 eV, 屬于放熱反應. 在勢能面上, 初態(分子吸附)和終態(解離吸附)之間的過渡態的吸附能為-0.561 eV, 如果從分子吸附狀態出發, 其反應活化能為1.159 eV; 而從解離狀態出發, 其反應活化能為1.270 eV, 也就是說在Cu(111)表面, 水分子分解反應的正方向的反應更容易進行, 所以在此表面上水分子以解離吸附為主要的存在狀態. 這說明對于水分子的裂解反應而言, Cu(111)表面具有較高反應活性.
圖2 水分子在Cu(111)表面上不同的吸附狀態與反應路徑, 及分子吸附和解離吸附的局部構型
Fig.2 Different adsorption states and reaction path of water on Cu(111) surface (a), and the local configurations of molecular adsorption (b) and dissociation adsorption (c)
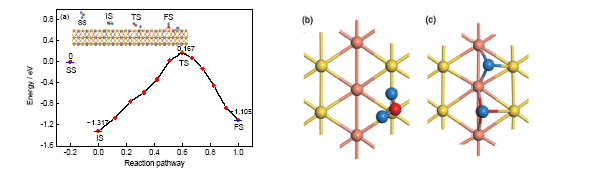
關于AuCu(111)表面的研究與應用已有諸多報道[25,26]. 本工作中分別優化了2種水分子可能的吸附構型, 即O原子位于Au原子上吸附(標記為AuCu(111)-Au), 以及O原子位于Cu原子上吸附(標記為AuCu(111)-Cu). 從圖3及表1可以看到, 對于AuCu(111)-Au模型中的分子吸附狀態, 水分子平面與表面接近于平行(夾角約為13.405°)吸附, 2個H原子分別指向鄰近的Au和Cu原子, 對應的距離分別0.3587和0.4757 nm. 分子吸附時的吸附能為1.317 eV. 在解離吸附狀態下, H+OH基團的平面與表面接近垂直(夾角約為83.432°), 裂解產物OH表面羥基豎直吸附于2個Cu原子之間的橋位, 另一裂解產物H原子吸附于由2個Cu原子和1個Au原子構成的空位. 解離吸附的吸附能為1.105 eV, 由此可見, 水分子在這個吸附位上的裂解反應是吸熱反應, 對應的反應熱為0.212 eV. 分子吸附和解離吸附之間的過渡態對應吸附能為0.167 eV, 其微觀結構接近于終態, 所以屬于“晚壘”過渡態. 從分子吸附狀態出發, 反應活化能為1.484 eV; 從解離狀態出發, 反應活化能為1.272 eV. 由于正反應活化能較高, 反應較難進行, 而且初態分子吸附的吸附能較高, 因此水分子在這個吸附位上以分子吸附為主要的存在形態. 分子吸附與解離吸附的局部示意圖如圖3b和c所示, O原子與其所吸附的Au原子的距離分別為0.2646 和0.2527 nm, 與最近鄰Cu之間的距離分別為0.3935, 0.4076 和0.1972 nm.
圖3 水分子在AuCu(111)-Au表面上不同的吸附狀態與反應路徑, 及分子吸附和解離吸附的局部構型
Fig.3 Different adsorption states and reaction path of water on AuCu(111)-Au surface (a), and the local configurations of molecular adsorption (b) and dissociation adsorption (c)
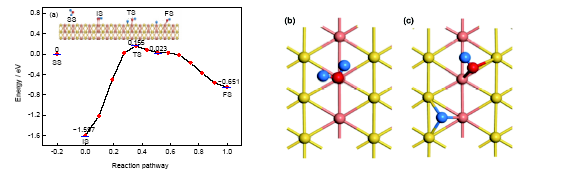
從圖4和表1可以看出, 對AuCu(111)-Cu模型, 分子吸附時水分子平面與表面的夾角約為20.408°, 過渡態時的夾角約為21.010°, 解離吸附時H+OH基團的平面與表面夾角約為22.745°, 可見水分子或者裂解產物的平面與表面的夾角在裂解前后的變化較小. 在裂解過程中, 其中1個H原子被活化之后與O原子分離, 并逐漸靠近表面, 最終吸附于由2個Cu原子和1個Au原子構成的空位; 同時O原子偏離頂位吸附, 與其相連的H原子則遠離表面(形成的表面羥基是傾斜在表面上的), 最終吸附于2個Cu原子之間的橋位. 從圖4a可以看出, 水分子在此吸附位上分子吸附和解離吸附對應吸附能分別是1.597和0.651 eV. 也就是說, 水分子在此吸附位上裂解反應的反應熱為0.946 eV, 是吸熱反應. 分子吸附和解離吸附之間過渡態對應的吸附能為0.155 eV, 其微觀結構接近于分子吸附狀態, 屬于“早壘”過渡態. 與以上其它3個反應路徑不同, 在這一反應路徑0.5附近有一個吸附能為0.023 eV的中間產物. 從總的反應路徑來看, 從分子吸附初態出發, 水分子需要躍過1.752 eV的活化能勢壘, 而其逆反應只需要0.806 eV. 這說明水分子在AuCu(111)的Cu位裂解時, 正反應較難進行, 所以在這一吸附位上水分子以分子吸附為主要的存在形狀. 分子吸附與解離吸附的局部示意圖如圖4b和c所示, O原子與其所吸附的Cu原子的距離分別為0.2136和0.1905 nm.
圖4 水分子在AuCu(111)-Cu表面上不同的吸附狀態與反應路徑, 及分子吸附和解離吸附的局部構型
Fig.4 Different adsorption states and reaction path of water on AuCu(111)-Cu surface (a), and the local configurations of molecular adsorption (b) and dissociation adsorption (c)
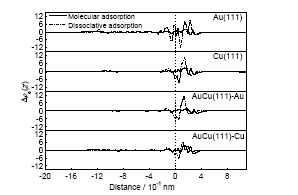
根據沿表面法線方向的平均電子密度變化(即平均差分電子密度曲線)可以分析在不同吸附狀態下水分子或者裂解產物與表面之間的電荷轉移情況. 從圖5可以看出, 對Au(111)面上水分子的分子吸附而言, 表面附近的電子密度減小, 說明有部分電子傳遞到了水分子上. 同時在水分子與表面之間的區域當中, 2種吸附狀態都是電子密度先增大后減小, 解離吸附對應的變化幅度更大. 對水分子或裂解產物而言, 其遠離表面一端的電子密度減小, 說明不論是分子吸附還是解離吸附, 電子均集中分布在O原子附近. 對Cu(111)表面的吸附而言, 水分子吸附之后, 表面的電子密度變化很小, 更主要的變化是水分子或者裂解產物內部電子的重新分布, 導致電子主要集中在O原子; 在解離吸附狀態下, 表面附近的電子大部分向水分子傳遞, 裂解產物中O原子位置附近的電子密度明顯增加, 說明電子集中分布在這個區域. 對于AuCu(111)-Au模型吸附, 分子吸附時表面的電子變化情況并不明顯, 而且表面上電子密度稍微有所增加; 解離吸附時表面上電子的情況出現先減小后增加、再減小的情況. 對于表面的電子變化很小的情況, 這可能主要是由于水分子內部電子的重新分布, 導致電子主要集中到O原子附近. 對于AuCu(111)-Cu模型吸附, 水分子吸附到表面之后, 表面電子密度略微增加, 說明在吸附過程中水分子的電子不僅有部分傳遞到表面上, 而且其內部也發生重組, 電子更加向其O原子靠近; 在解離吸附狀態下, 電子先減小再增加, 而在分子吸附時則是一直增加. 可見對于分子吸附, 由于其與表面作用強于解離吸附, 分子吸附狀態應該更加穩定. 綜上所述, 電子轉移的計算結果表明, 轉移的電子數越多, 意味著吸附物與表面的相互作用越強, 因此在分子吸附時Au(111)表面的吸附能最大, 而在解離吸附時AuCu(111)-Au具有最大的吸附能. 上述電子轉移的計算結果說明了2.1節中不同吸附狀態的能量關系.
圖5 水分子在Au(111), Cu(111), AuCu(111)不同吸附狀態沿表面法線方向的平均差分電子密度圖
Fig.5 Profile of mean electron density difference of water on Au(111), Cu(111), AuCu(111) surfaces by the different adsorption states
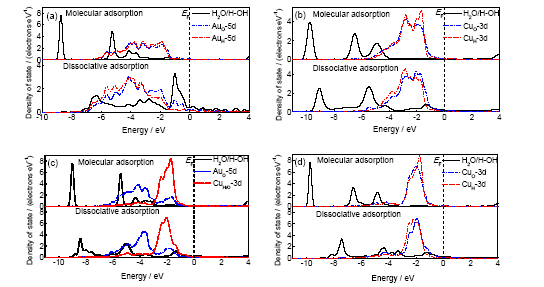
從電子結構的變化可以進一步說明水分子或者裂解產物在表面上吸附的異同, 以及它們之間的能量對比關系. 從圖6可以看出, 在4種表面上的分子吸附時, 水分子的1b2電子態與孤立水分子是一致的, 水分子與表面的相互作用主要是通過其孤對電子態(包括3a1和1b1)與Au或Cu的d電子態相互雜化而發生作用. 但是在Au(111)表面和AuCu(111)-Au表面上水分子的3a1電子態與孤立水分子相比變化很小, 而在Cu(111)表面和AuCu(111)-Cu表面上時則變化較為明顯. 同時水分子的1b1電子態在Au(111)表面和AuCu(111)-Au表面上完全與Au的d電子態重疊, 發生明顯的雜化, 而在Cu(111)表面和AuCu(111)-Cu表面上時則變化較小. 另一方面, Cu的d電子態與Au的d電子態相比, 更容易形成靠近費米能級的孤立電子態. 這些計算結果說明, 對于分子吸附狀態, Au與水分子的相互作用比Cu與水分子的相互作用更強; 對于解離吸附狀態, H+OH基團的類1b2電子態在Au(111)表面上完全消失, 并且與3a1電子態一起與Au的d電子態完全重疊、雜化, 但是在Fermi能級EF附近有一個未完全雜化的類1b1電子態, 該電子態主要由頂位吸附的表面羥基形成, 與吸附位上的Au原子有一定的相互作用. 在其余3個表面上, H+OH基團表現出一定的類1b2和3a1電子態特征, 而其類1b1電子態則與吸附位上的d電子態完全重疊、雜化. 從另一方面來看, 由于Cu的d電子態比Au的d電子態更靠近Fermi能級, 根據金屬催化材料的d帶中心理論, 即Cu比Au具有更強的催化活性, 而且在有Cu存在的表面上, 水分子的1b1電子態或者H+OH基團的類1b1電子態都發生更為完全的重疊和更明顯的雜化. 從這個角度可以解釋在AuCu(111)表面上, 水分子裂解反應的反應能更小, 甚至在Cu(111)表面上解離吸附比分子吸附更穩定, 水分子的裂解反應成為放熱反應. 這一變化的內在原因就在于水分子或H+OH基團與表面金屬原子不同類型的相互作用, 以及金屬原子d電子態所反映的活性不同.
圖6 水分子在不同表面上以分子吸附或解離吸附時的局域分波態密度圖
Fig.6 Local and partial density of states of water on Au(111) surface (a), Cu(111) surface (b), AuCu(111)-Au surface (c) and AuCu(111)-Cu surface (d) at the present of molecular adsorption or dissociation adsorption (EF—Fermi level)
(1) 在從分子吸附到解離吸附的反應路徑上, 以反應活化能為比較標準, 水分子在所考慮的4種模型中的反應活性順序如下: Au(111)<AuCu(111)-Cu<AuCu(111)-Au<Cu(111). 這與水分子的分子吸附狀態在Au(111)表面和AuCu(111)-Cu表面具有相對較大的吸附能, 而解離吸附狀態具有相對較小的吸附能有關. 與此相反的是, 水分子的分子吸附狀態雖然在AuCu(111)-Au表面和Cu(111)表面具有相對較小的吸附能, 但同時解離吸附狀態時的吸附能也相對較大.
(2) 從不同吸附狀態與表面電子轉移情況看, 兩者之間電子轉移較多, 則意味著相互作用較強, 對應的吸附能較大, 所以分子吸附時Au(111)表面的吸附能最大, 而解離吸附時AuCu(111)-Au的吸附能最大.
(3) 對于水分子在金屬催化劑表面的裂解反應, 過渡金屬Cu具有更高的活性, 因此在實際反應體系中, 用AuCu二元金屬合金替代貴金屬Au催化劑不僅可以降低材料成本, 還可以提高反應活性.

1 計算方法和模型
2 結果與討論
2.1 分解反應路徑
Model
State
Au(111)
IS
0.0984
0.0985
104.587
0.3276
0.3185
0.2571
4.375
TS
0.0983
0.1883
154.758
0.3768
0.1641
0.2125
78.141
FS
0.0986
0.2237
153.875
0.3664
0.1619
0.2113
57.029
Cu(111)
IS
0.0988
0.0988
105.354
0.2857
0.2858
0.2168
15.190
TS
0.0987
0.1569
126.960
0.2778
0.1773
0.1968
41.649
FS
0.0981
0.3866
95.969
0.2731
0.1735
0.1997
88.792
AuCu(111)-Au
IS
0.0983
0.0985
105.055
0.3646
0.3135
0.2646
13.405
TS
0.0986
0.1503
141.136
0.2922a
0.1839a
0.2310a
38.957
0.2992b
0.1915b
0.2314b
FS
0.0982
0.3077
95.809
0.1972a
0.1762a
0.1745a
83.432
0.2527b
0.1727b
0.1874b
AuCu(111)-Cu
IS
0.0987
0.0988
105.907
0.3226
0.3053
0.2136
20.408
TS
0.0975
0.2211
127.245
0.2931a
0.1624a
0.1852a
21.010
0.3097b
0.2483b
0.2997b
FS
0.0987
0.4212
114.274
0.2587a
0.1914a
0.1905a
22.745
0.2872b
0.1755b
0.2328b
2.2 差分電子密度
2.3 態密度
3 結論
來源--金屬學報
 滬公網安備31011202020290號
滬公網安備31011202020290號
